

Prof Jill Clayton-Smith discusses the genetic underpinnings of Angelman syndrome
Genetics of Angelman Syndrome
Visit our key topics section and select 'genetics' from the drop down menu to read a general introduction about genetics .
Angelman syndrome is caused by missing or altered genetic information on the maternal copy of chromosome 15. We inherit two copies of each chromosome one from each parent and have 46 chromosomes in total.
The specific region of chromosome 15 affected in Angelman syndrome is called 15q11.2-q13. This simply locates the missing genetic information to a particular area on the chromosome.
It is important to remember that Angelman syndrome arises when the genetic information on 15q11.2-q13 on the maternal (mother's) copy of the chromosome is missing or altered. Missing genetic information in the same region of the father's copy of the chromosome results in another genetic syndrome called Prader-Willi syndrome. This parent-specific gene activity (expression) is called genomic imprinting.
The genetic alterations of chromosome from 15q11.2-q13 that results in Angelman syndrome can arise in four different ways. These four genetic mechanisms are often described as genetic subtypes of Angelman syndrome:
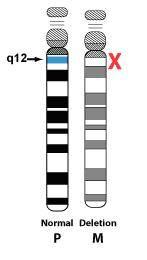
Deletion:
Approximately 70% of individuals with Angelman syndrome have a deletion of the mother's copy of 15q11.2-q13. This means that the information is simply missing. Most deletions are of a similar size, but when examined in detail they can be divided into two groups, with Class 1 deletions being just slightly larger than Class 2 deletions.
Paternal Uniparetnal Disomy (UPD):
Approximately 7% of individuals with Angelman syndrome have a uniparental disomy which means that both copies of chromosome 15 have been inherited from the father and so the mother’s copy is missing.
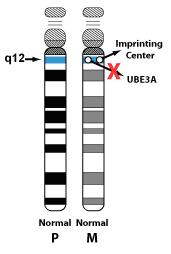
Imprinting Centre Defect:
Approximately 3% of individuals with Angelman syndrome have an imprinting centre defect in the 15q11-q13 region. This means that the individual has inherited a copy of chromosome 15 from each parent but the maternal copy is not expressed as it should be.
UBE3A Mutation:
Approximately 11% of individuals with Angelman syndrome have a mutation of the UBE3A gene which lies within the 15q11-q13 region and is the critical gene for Angelman syndrome. UBE3A has several functions including brain development and involvement in protein turnover in the brain. This loss of UBE3A function causes the characteristic features of Angelman syndrome.
Clinical Diagnosis:
This subtype refers to individuals who have received a clinical diagnosis (a diagnosis based on physical features and behaviour) but after having blood tests are found to have no genetic difference (i.e. negative blood results for the above genetic causes). Most of these individuals will not have Angelman syndrome but will have other similar and overlapping disorders, which should be considered during diagnosis. It is possible, however, that there may be other genes involved in Angelman syndrome and research is ongoing to try and discover more.
Prof Jill Clayton-Smith discusses genetic counselling in the following video:
For further information about genetic counselling in Angelman syndrome we recommend you visit